|
|
||
10/07/10 |
|
|
A very fine example of a secondary metabolite gene cluster is the one responsible for the production of the red/yellow pigment aurofusarin, which we have analyzed extensively and still work on. The following text is mainly taken from my master thesis, but has been updated to include data we have obtained later. The aim is to describe the process of determining the biosynthetic pathway for af a secondary metabolite (polyketide).
Aurofusarin (mycelium pigment)Aurofusarin was first described as a Fusarium culmorum pigment by Ashley et al. (1937) during a larger project to characterise the metabolites of filamentous fungi (Booth 1971). The colour of aurofusarin is dependent on the pH value of the solvent, ranging from golden yellow in acidic solvents to red/purple in alkaline solvents (Ashley et al. 1937). The pigment is produced in high quantities, continuously during mycelium development, resulting in the increasing staining of both mycelium and medium (the mycelium shifts from white to yellow and finally to a deep red colour). During determination of the structure of the naphthopyrone rubrofusarin, which resembles the aurofusarin monomer, it was noted that culture conditions (stir and no stir) affected the production of aurofusarin and rubrofusarin in reverse complementary (Leeper & Staunton 1984).
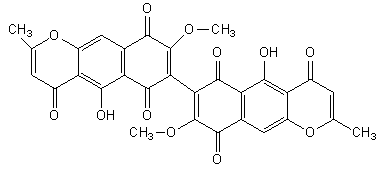
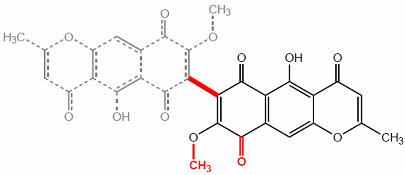
Figure 1 The structure of aurofusarin
Later investigations have shown that aurofusarin (C30H18O12) is a homodimeric naphthoquinone, with a molecular mass of 570 g/mol (Figure 1). Known producers include Hypomyces rosellus, Dactylium dendroides, Fusarium acuminatum, F. avenaceum, F. crookwellens, F. culmorum, F. graminearum, F. poae, F. pseudograminearum, F. sambucinum, F. sporotrichioides and F. tricinctum (Samson et al. 2000). Aurofusarin is produced under various sub-optimal conditions, such as high or low pH, high temperatures and phosphate starvation. The ambient pH is the most significant, as many naphthoquinones are cytostatic at neutral pH (Medentsev & Akimenko 1998). The genes responsible for the production of aurofusarin could be regulated by a global pH regulatory factor, such as PacC in Aspergillus sp., but this is still unknown.
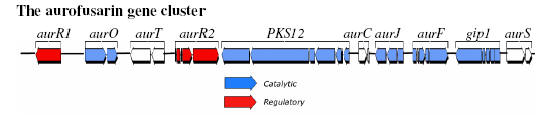
The aurofusarin gene cluster in Fusarium graminearum The aurofusarin gene cluster was first described by Malz et al. (2005), which represent a collaboration between our group (headed by Professor Henriette Giese) and Professor Wilhelm Schäfers group at the University of Hamburg (Germany). The gene cluster was identified by random Agrobacterium tumefaciens mediated mutagenesis followed by isolation of transformants affected in the aurofusarin biosynthesis (those with a white or yellow phenotype). Mapping of T-DNA integration sites in the isolated transformants showed that four independent integrations had occurred in the promoter region between AurR1 and AurO. A bioinformatic analysis of the surrounding genes showed that it included a polyketide synthase gene and several genes encoding enzymes that possibly could participate in the biosynthesis of aurofusarin (Table 1). The bioinformatic analysis also showed that AurR1 most likely was a pathway specific transcription factor, of the binuclear zinc cluster class, responsible for expression from the genes in the cluster (Figure 1). Reverse transcriptase PCR of the random mutants showed that the surrounding genes were not expressed.
Figure 2 The aurofusarin gene cluster in F. graminearum, located on contig 1.116 as defined by Malz et al. (2005).
Targeted disruption of PKS12 followed by HPLC analysis of the mutant showed that this polyketide synthase was required for aurofusarin production (Malz et al. 2005). Deletion of gip1, a laccase, showed that this enzyme also was required for aurofusarin biosynthesis (Kim et al. 2005). A microsynteny analysis of F. graminearum, F. culmorum and F. pseudograminearum revealed that the proposed gene cluster is conserved across these species (Malz et al. 2005).
Table 1 Results of the bioinformatic analysis of the aurofusarin gene cluster by Malz et al. 2005, similar results were obtained by Kim et al. 2006.
Transcriptional regulation of the aurofusarin gene cluster The subject of my master thesis was the transcriptional regulation of the aurofusarin gene cluster. The analysis by both Malz et al. 2005 and Kim et al. 2006 predicted that the cluster contained three genes (aurR1, aurR2 and aurJ) that could be involved in the transcriptional regulation of the gene cluster. AurR1 and aurR2 contains binuclear zinc clusters (Zn2Cys6), typical of fungal transcription factors, such as Gal4. The three genes were replaced by targeted Agrobacterium tumefaciens mediated transformation using the pAg1-H3 vector system, described by Zhang et al. 2003. The effects of the deletions on visible phenotype and chemotype are shown in table 2.
Table 2 Effects of targeted deletion of genes suspected of being involved in regulation. For the visual phenotype the strains were grown on Defined Fusarium Medium.
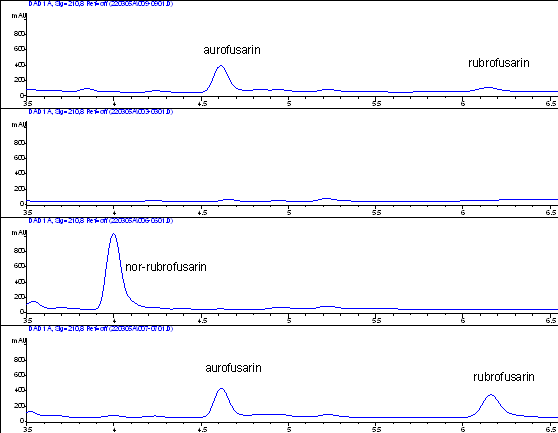
The HPLC-UV analysis showed that the ∆aurR1 mutant did not produce rubrofusarin nor aurofusarin, indicating that required biosynthetic apparatus was not present in the mutant. The ∆aurJ mutant also did not produce aurofusarin or rubrofusarin, but accumulated a new metabolite, which by 1D og 2D 1H-NMR and 13C-NMR was determined to be nor-rubrofusarin. The ∆aurR2 mutant produced aurofusarin in levels similar to those found in the wild type, however its production of rubrofusarin was significantly increased compared to the wild type. To directly determine the effects on transcription of genes in the gene cluster, the expression was analysed by RT-PCR (see figure 3).
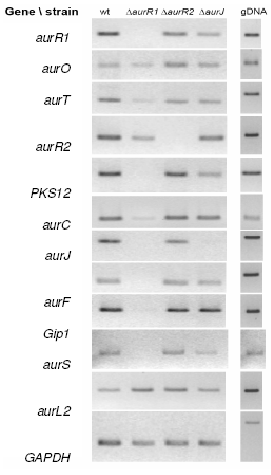
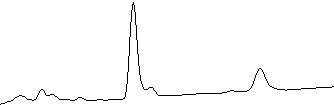
Figure 3: Reverse transcriptase PCR analysis of the aurofusarin gene cluster in three different deletion mutants (∆aurR1, ∆aurR2 and ∆aurJ) and the wild type. The analysis showed that no detectable changes had occured in the ∆aurJ and ∆aurR2 mutants. In the ∆aurR1 mutant no transcript of PKS12, aurJ, aurF, gip1 and aurS genes could be detected, while the expression of aurT, aurO and aurC also were negatively affected. This shows that AurR1 is a positive transcription factor that is required for the expression of the aurofusarin gene cluster. Unproven theory: AurR2 may function as a co-activator to AurR1, fine tuning the expression from the gene cluster, allowing a controlled switch between biosynthesis of rubrofusarin and aurofusarin.
Theories for the biosynthetic pathway of aurofusarin
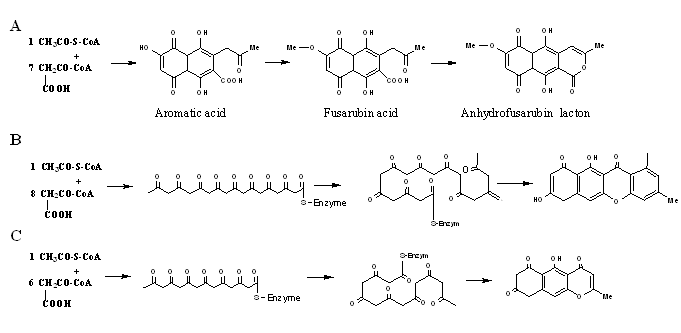
The folding pattern utilized during biosynthesis of aurofusarin and related naphthoquinones Aurofusarin is a homodimeric naphthoquinone and it is reasonable to assume that the two monomers are synthesized independently and then subjected to dimerisation. The biosynthesis pathway for the naphthoquinones dihydrofusarubin, fusarubin, javanicin and solaniol (all naphthazarins) in Fusarium has been summarized by Medentsev & Akimenko (1998). The first stable intermediate, in the Medentsev & Akimenko pathway, following the action of the PKS is aromatic acid, which contains two intramolecular cyclization bonds between position C2/C11 and C4/C9 (Figure 4). However, it is not possible to convert aromatic acid into the aurofusarin monomers, as the third intramolecular cyclization reaction results in a wrong positioning of the ether bond within the ring (exemplified by fusarubinic acid to anhydrofusarubin lacton in Figure 4A).
Figure 4 A) Part of the biosynthesis mechanism of naphthoquinones presented by Medentsev and Akimenko (1998). B) The Yadav et al. (2005) folding scheme for the pre-naphthoquinone bikaverin. C) The folding scheme that has been experimentally proven true for the naphthopyrone rubrofusarin. This scheme may also be true for the aurofusarin monomer and bikaverin.
This problem has theoretically been solved by Yadav et al. (2005) for bikaverin which resembles the aurofusarin monomer (Figure 4B). Their folding scheme is entirely plausible with respect to the positioning of the ether bond and could also be true for the aurofusarin monomer. However, this is not the only possible solution to the problem (Figure 4C), as pointed out by Leeper & Staunton (1984) in their investigation of the naphthopyrone rubrofusarin, produced by F. graminearum and F. culmorum. The folding scheme shown in Figure 4C and Figure 5 will also result in the same final product. 13C NMR analysis of rubrofusarin has shown that the second folding scheme is used in vivo. A similar study with the naphthopyrone WA performed by Isao et al. (2001) gave similar results. The PKS responsible for the biosynthesis of WA has been characterized, while this is not the case for rubrofusarin.
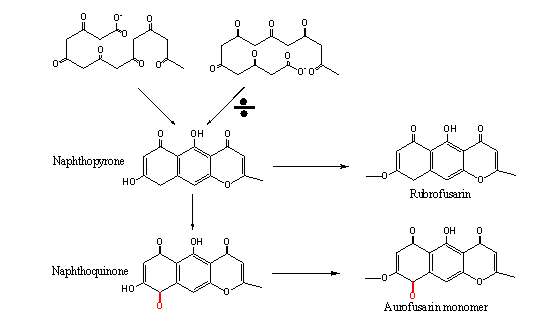
Figure 5 Top: Two folding patterns resulting in the same product, the pattern to the left has been shown to be the one utilized in vivo by PKSs. Bottom: The only difference between naphthopyrones (rubrofusarin/WA type) and naphthoquinones (aurofusarin type) is the second ketone group found in the first ring (highlighted with red).
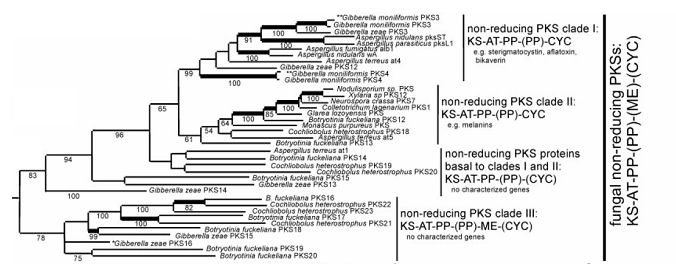
A biosynthesis link between naphthoquinones and naphthopyrones The aurofusarin monomer and rubrofusarin belong to different chemical classes, the naphthoquinones and naphthopyrones respectively (naphthopyrones are characterized by the ketone group located complementary to the inter-cyclic ether bond). The two compounds only differ with respect to the second ketone group found on the first ring of aurofusarin (Figure 5, highlighted ketone group). This ketone group, which defines the naphthoquinones, is also found in the compounds described by the Medentsev & Akimenko (1998) biosynthesis pathway (left in Figure 4A). However, while the rest of the oxygen groups can be traced back to the original unfolded polyketide, the Medentsev & Akimenko pathway does not explain the origin of the second ketone group, and it must therefore be the result of post-PKS modifications.does not explain its origin, The first stable intermediate in the biosynthesis of anhydrofusarin lacton is therefore not the naphthoquinone Aromatic acid, but the corresponding naphthopyrone. This must also be true for all other naphthoquinones, as no folding scheme will result in positioning of two ketone groups on adjoining carbon atoms, if all extender units during the synthesis have been malonyl-CoA (Figure 5). The established link between naphthoquinones and naphthopyrones places them within the same biosynthesis pathway, with naphthopyrones being precursors for naphthoquinones. This opens for the possibility that the involved PKSs, gene clusters and biosynthesis pathways share a closely linked evolutionary history, as only one or two enzymatic steps (catalysing the addition of oxygen) has to be acquired to convert a naphthopyrone gene cluster/biosynthesis pathway into a naphthoquinone gene cluster/biosynthesis pathway. Phylogenetic analysis support this as the corresponding PKSs (pks12 (aurofusarin), pks4 (bikaverin) and WApks (WA)) cluster tightly together, as members of the sub-class “non-reducing PKS clade I” (Kroken et al. 2003). This sub-class belongs to the class of “fungal non-reducing PKSs”, characterized by the presence of the Claisen-cyclase domain and the absence of the methyl transferase domain (Figure 6), corresponding to the WA type PKS defined by Bingle et al. (1999). pks1 responsible for melanin biosynthesis (belonging to non-reducing PKS clade II) in A. fumigatus has been shown to follow the same folding scheme as described for WA (Tsai et al. 2001). It is therefore possible that this scheme is true for all polyketides produced by PKSs belonging to the non-reducing PKS Clade I and II, based on the shared Claisen-cyclase domain. The exception is the PKSs responsible for synthesis of aflatoxin and sterigmatocystin (pksST), as these enzymes use alternative starter units, synthesized by Fas1 and Fas2 (see the page on aflatoxin).
Figure 6 Phylogenetic relationship between fungal PKSs belonging to the non-reducing subclass, as defined by Kroken et al. (2003). The figure is part of a larger phylogeny taken from Kroken et al. (2003).
The folding scheme for the aurofusarin monomer most likely resembles the one described for rubrofusarin and WA rather than the one described for other naphthoquinones. This is based on the structural similarity and differences between the compounds and the close evolutionary relationship between the responsible PKSs (pks12 (aurofusarin) and WApks (WA). Known rubrofusarin producing fungi includes F. culmorum and F. graminearum, which also produces aurofusarin. During determination of the folding scheme for rubrofusarin (Leeper & Staunton 1984) it was observed that simple changes in culture conditions resulted in a switch between production of rubrofusarin and aurofusarin. It is very likely that the two compounds are part of the same biosynthesis pathway with rubrofusarin being the precursor of the aurofusarin monomer. A consequence of this is that PKS12, which has been shown to be the originator of aurofusarin (Malz et al. 2005), also is responsible for the biosynthesis of rubrofusarin. This is further supported by the observation that the replacement of aurR2 results in a changed ratio between aurofusarin and rubrofusarin compared to the wild type, and that replacement of aurR1 results in a lack of both aurofusarin and rubrofusarin biosynthesis (see table 2).
Prediction of the enzymes required for the biosynthesis of aurofusarin Question: Is it possible to predict which enzyme classes are involved in the biosynthesis of aurofusarin, by analysing the structure of final product? Aurofusarin is a homodimeric polyketide. Now if we assume that the two identical portions of the molecule is synthesised independently by the same enzymes and then joined together, or that the same tailoring enzymes can form the identical groups in each end of the molecule, we have already reduced the number of required enzymes by 50 %. By tracking the polyketide backbone of the molecule it should be possible to identify which groups are the direct result of the polyketide synthase and which have to be added later by tailoring enzymes. This form of reverse engineering is possible when dealing with the products of non-reducing polyketide synthases, as their products will be unmodified polyketide chains (-(CH2-CO)n-). The polyketide synthase, PKS12, that catalyses the formation of aurofusarin is a minimal polyketide (Figure 7), with a C'-terminal Claisen cyclase domain. The presence of a Claisen cyclase domain suggests that the product of this PKS will have an aromatic form. The aromatic structure is formed by aldol type condensation between an internal keton/enol and a -CH2- group in a lineary polyketide, resulting in formation of a double bond and a ring closure.
Figure 7 The domain structure of PKS12. KS = b-ketoacyl synthase , AT = Acyltransferase, ACP = Acyl Carrier protein and CYC = Claisen cyclase. The prediction was made using the http://www.nii.res.in/nrps-pks.html prediction program.
Figure 8 Overlay of the folding pattern of nor-rubrofusarin onto the structure of aurofusarin. The non-shared groups are highlighted with red and includes a ketone group, a methoxy group and the dimerization bond. The two possible folding patterns that were shown in figure 5, results in identical locations of side groups (hydroxy and methyl groups), meaning that the following analysis will be independent of which of the folding patterns are truly used in vivo. By overlaying the folding pattern onto the structure of the aurofusarin monomer, it becomes apparent that the methoxy group (-O-CH3), the ketone group and the dimerisation bond are not the direct product of the polyketide synthase, but have to be added by tailoring enzymes. Methoxy groups are typically formed by O-methyltransferase. Ketone groups can be formed by a two step oxidation process: 1) hydroxylation catalyzed by either a laccase or a monooxygenase and 2) oxidation of the formed hydroxy group by an oxidoreductase resulting in formation of a ketone. Both laccaes and monooxygenases have been proposed to catalyse the dimerisation of polyketides
First proposal for a biosynthesis mechanism for the aurofusarin monomer (NOT CORRECT!!) Numbering of carbon atoms in the following section does not follow the standard IUPAC nomenclature, but is based on the order in which the C-atoms have been added to the polyketide chain (oldest carbons recieves highest number).
Figure 9 Numbering of carbon atoms and rings in the aurofusarin monomer. The condensation of 1 acetyl-CoA and 6 malonyl-CoA units by the PKS results in formation of a heptaketide. The heptaketide undergoes Claisen- and Aldol-type cyclization reactions following the rubrofusarin/WA folding scheme as described in (Figure 5), resulting in formation of two carbon-carbon bonds and one ether bond (Figure 10B). The C1 ketone group can undergo enol-keto tautomarization, resulting in formation of a double bond between C1 and C10, as part of a conjugated bond system within ring B. In a similar fashion the C5 ketone group changes from the keto to the enol tautomer (C4-C5 double bond), driven by the coupling to the C2-ketone group and conjugated bond system in ring B (Figure 10D-E). This will allow a Laccase(Cu2+) to react with the formed alcohol on C5, resulting in deprotonation of the alcohol (+ Laccase(Cu+) + H+) and creation of a free radical (Figure 10E-F). The created free radical can be reversed by “stealing” an electron from the double bond found between C4 and C5, leading to formation of a new C5 ketone group. The result of this electron relocation is that the free radical is transferred from the former alcohol group to the C4 carbon atom (Figure 10F-G)(the laccase or monooxygenase could also attack the C1 or C3 hydroxyl group as this could yield the same result via the conjugated bond system). By allowing two of these reactive pre-aurofusarin monomers to react, a C-C bond can be formed between the C4 atoms on each of the monomers (Figure 10G-H). The two C5 ketone groups can then undergo enol-keto tautomarization, driven by the formation of conjugated double bonds between C2-C7, C2-ketone group and C4-C5 on the opposite aurofusarin monomer (Figure 10H-I). The C5 alcohol on each of the monomers can then undergo methylation (Figure 10J), most likely with S-adenosyl-L-methionine as donor, as described for other polyketide systems such as javanicin (Gatenback & Bentley 1965). The ketone group found on C6 of the final aurofusarin monomer are most likely formed by two independent enzymatic reactions. First a monooxygenase introduces a hydroxyl group (Kaim & Schwederski 2001). This group can then be further oxidized by a dehydrogenase (also known as oxidoreductases) to form the final ketone group (Figure 10K and L).
Figure 10 First proposal for a biosynthesis mechanism for aurofusarin in Fusarium sp.. (A) The unmodified heptaketide produced by PKS12. (B-E) Enol-keto tautomer transition. (F) Laccase deprotonates the C5 hydroxyl group, resulting in formation of a free radical on C4 (G). Dimerization of two pre-aurofusarin monomers (H) and formation of conjugated bonds. (J) Methylation of the C5 hydroxyl. (K) Introduction of a hydroxyl group on C6 and (L) its further oxidation to form a ketone group. Note that step J, K and L are not stoichiometrically balanced.
It is at present not possible to theoretically determine the exact order of the modification steps (dimerisation, methylation, oxidation and dehydration). In addition, step D to G can also work with the C6-H2 group, ultimately resulting in formation of a free radical on C6 instead of C4, this mechanism can explain other dimerisation patterns (see later). Rubrofusarin can be formed by A-D and J omitting step F-H and K-L. This model was not correct, but formed the basis for our further work. When it was combined with data from the targeted gene replacement experiments it was possible to narrow down the number of possible biosynthetic pathways (determine the order of reactions).
Targeted replacement of the genes in the aurofusarin gene cluster The proposed biosynthetic pathway (figure 10) put forth a lot of ideas and raised a lot of questions, which could only be anwesered through experimental work. With the aim of determining whether the co-regulated genes in the aurofusarin gene cluster were involved in the biosynthesis and to determine which reactions they catalyzed, I knocked out 6 additional genes (aurT, aurC, aurF, GIP1, aurS, aurL2) by Agrobacterium tumefaciens mediated transformation. The resulting mutants displayed very destinct colour phenotypes which were supported by the follwing HPLC-DAD analysis (figure 11).
Figure 11 Selected aurofusarin mutants.
Chemical characterization of the accumulating metabolites in the individual mutants, performed by PhD-stud. Nikoline Nielsen, showed that several of the mutants accumulated the same compound; rubrofusarin (Table 3).
Table 3
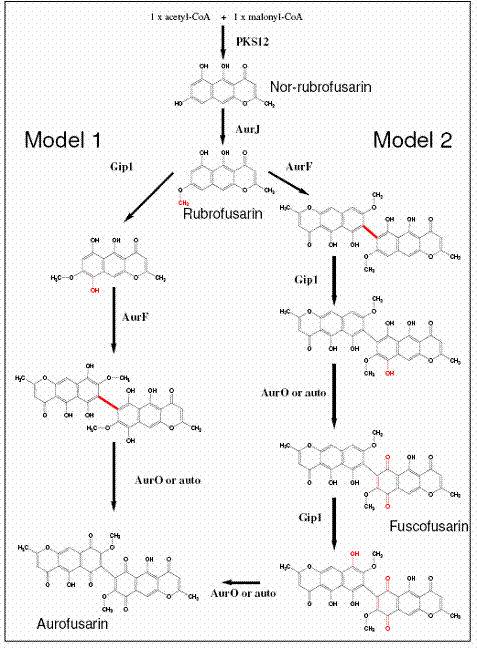
The analysis showed that the PKS12, aurJ, aurF, GIP1, aurC and aurS mutants all were impaired in aurofusarin biosynthesis, while aurO and aurT mutants displayed an altered aurofusarin/rubrofusarin ratio. Based on this data it became apparent that AurJ (O-methyltransferase) was the first tailoring enzyme in the biosynthesis pathway, catalysing the conversion of nor-rubrofusarin to rubrofusarin by a O-methylation of the C5-OH group to C5-O-CH3. Rubrofusarin is then processed by aurF, GIP1 and aurO to form aurofusarin, however based on the accumulation of rubrofusarin in these three mutants it was unfortunately impossible to directly determine which reaction the individual enzymes catalyze. Both AurF (monooxygenase) and GIP1 (laccase) could potentially catalyze the dimerization reaction or oxidation of C6, while aurO can "only" be responsible for oxidizing the C6 hydroxy group to a ketone group. Based on this we end up with two possible biosynthetic pathways (Figure 12)
Figure 12 The two possible biosynthetic pathways for aurofusarin.
Several of the mutants surprisingly accumulated rubrofusarin, and not as expected different intermediats. A first obvious explanation for this could be that the compounds downstream of rubrofusarin is unstable and have degraded, or that they are toxic to the producing cells thus limiting their production. However, we now believe that some of the proteins form a protein complex, and that removal of one component results in a complete loss of function for all enzymes in the complex (unproven - but we are working on it).
Compounds related to aurofusarin (PubChem database search) At the The National Institutes of Health (USA) Substance and Compound database hosted at the PubChem server (http://pubchem.ncbi.nlm.nih.gov/) it is possible, based on Chemical structures, to search for compounds which displays similar structures and sub-structures as the query compound. This facility was “discovered” late in the project but the obtained results confirms the theoretical work on the folding scheme and biosynthesis pathway. A search with the aurofusarin structure resulted in > 200 hits. Manual inspection of these led to the identification of 16 compounds which shared the same ring structure and positioning of the internal ether bond (Table 3). The Medentsev & Akimenko (1998) paper contains a list of naphthoquinones of different ketide length. Ten of these have the same ether bond as found in aurofusarin (6-methyl-dehydro-a-lapachone, kanarion, rodocladonic acid, cryptosporin, funacin, lambertellin, erythrostominone, deoxyerythrostominone, epierythrostominol and fuscofusarin (dimer).
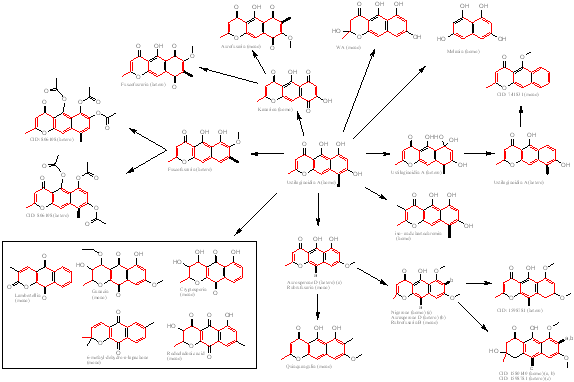
Table 3 Distribution of identified structures which display similarity to aurofusarin. Many of the identified polyketides (all heptaketides) can be formed via the proposed biosynthesis mechanism for aurofusarin through very few enzymatic steps (Figure 11).
Figure 11 Relationship diagram for the identified heptaketides showing similarity to aurofusarin. Dimeric compounds have been broken up into monomers. Highlighted bonds represent dimerisation bonds between monomers. For monomers that are found in multiple dimeric compounds with different dimerisation patterns, the dimerisation bond is marked with a letter. The identified polyketides have been isolated from many different species and the biosynthesis pathway shown in the figure cannot be found in one single fungal specie. The box represents a group of naphthoquinones with the same ester bond as seen in aurofusarin, but with the defining ketone groups found on ring b instead of c. The monomers that make up Ustilaginoidin A (center) represent the unmodified heptaketide folded via the rubrofusarin/WA folding scheme.
Dimerisation patternsThe identified dimeric naphthoquinones include both homo- and heterodimeric compounds. The dimerisation pattern of these were analysed, and both dimerisation between similar and different carbon atoms were identified. The analysis showed that three different carbon atoms (C4, C6 and C8) can participate in dimerisation bonds (Figure 12).
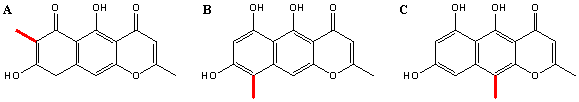
Figure 12 Possible dimerisation bonds (red). Type A) found in aurofusarin, B) in chaetochromin and C) in nigerone.
The three different dimerisation modes can be achieved through the same radical producing lacasse mechanism proposed for aurofusarin, working on either the C1, C3 or C5 hydroxyl groups. Mode C is possible due to the conjugated bond system, which allows free radicals to travel from the initial activated carbon atom (C4 or C6) to C8, resulting in its activation.
Additional reading:
References
|
Dette sted blev sidst opdateret 10. July 2010