|
|
||
01/08/10 |
|
|
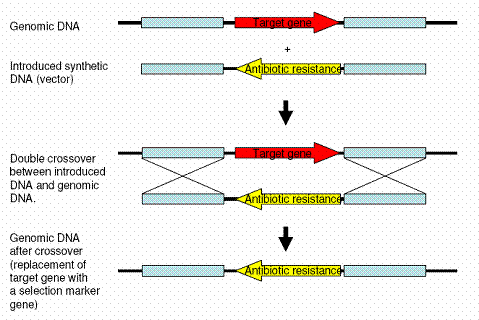
Functional genomics in fungiThe functional characterization of genes is typically dependent on the isolation or construction of mutants. In fungi specific mutants are typically constructed by a process known as targeted gene replacement by homologous recombination. Homologous recombination is a natural DNA repair mechanism that deals with double stranded DNA breaks during replication, it is believed to exist in all eukaryotes but is exceptional active in fungi. In homologous recombination two identical DNA pieces are combined to form a single continuous piece of DNA, restoring the sequence that existed prior to the double strand DNA break. In targeted gene replacement this process is utilized to combine introduced DNA pieces with the genomic target. The introduced DNA piece consists of a selection marker gene, typically a gene that codes for resistance to an antibiotic compound, surrounded by two DNA sequences that are identical to the sequences surrounding the genomic target (see figure).
FIGURE: Targeted gene replacement: Red arrow: the gene that has to be replaced (knocked out), Yellow arrow: the selection marker gene, in this case an antibiotic resistance gene, Blue boxes: Homologous recombination sequences, typically between 0.7 and 2 kb long.
Correct replacement mutants can then be isolated by culturing the fungi in the presence of the antibiotic compound, which will kill fungi that have not received the antibiotic resistance gene. The process of targeted gene replacement can be divided into the following steps:
Construction of the replacement vector has until recently been a fairly complex process, requiring two subsequent cloning steps to introduce the two homologous recombination sequences onto either site of the antibiotic resistance gene in the vector. STEP 1: Identification of target geneTarget genes can be identified by many different techniques depending on the aim of the specific study. The publication of the full Fusarium graminearum genome means that in today's research most target genes are identified through different bioinformatic techniques, such as Blast or co-expression. However, truly ground breaking discoveries are typically generated by screening of random mutant libraries for the loss or gain of a particular property
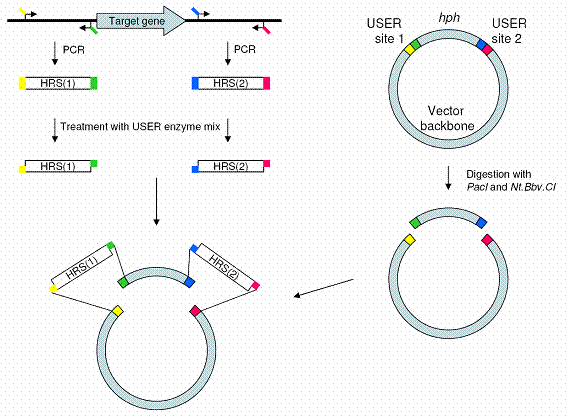
STEP 2: Construction of vectors for targeted gene replacementConstruction of the replacement vector has until recently been a fairly complex process, requiring two subsequent cloning steps to introduce the two homologous recombination sequences onto either site of the antibiotic resistance gene in the vector. We have developed a new vector system that allows for single-step construction of replacement vectors, a process that normally requires at least two cloning steps. The new vector construction strategy has reduced the construction time from ten to only three days, and reduced the work load by half compared to previous strategies. The simultaneous cloning of the two required PCR amplicons also gives a higher degree of freedom, regarding which portion of the target gene that should be replaced.
The new vector system and construction strategy is dependent on USER friendlyTM cloning, which in its commercial version provides highly efficient directional cloning of single PCR amplicons. However, we have found that the USER FriendlyTM cloning technique also is capable of fusing two PCR amplicons and two vector DNA pieces together in a single cloning step. The directional four fragment cloning has an average efficiency of 85 % (Rasmus J.N. Frandsen et al. 2008). This new strategy should make the more traditional gene disruption strategies obsolete.
Figure: The USER friendly cloning strategy we utilized for one-step construction of targeted replacement vectors. The two PCR amplified inserts are mixed with the two vector pieces and treated with the USER enzyme mix, which creates 9 bp long compatible overhangs. The mix is then transformed into E. coli cells, which catalyses the formation of covalent bonds between the DNA pieces. This four fragment cloning has an efficiency of 85 %, which is much higher than what can be achieved by standard cloning techniques, such as restriction enzyme and ligase dependent cloning, Xi-cloning or In-Fusion cloning.
STEP 3: Introduction of the vector into the fungusThe process of introducing DNA into the fungus is called transformation. Many different transformation techniques have been developed, such as biobalistics, electroporation, protoplast and Agrobacterium tumefaciens mediated transformation (ATMT). In our laboratory we utilize both protoplast and Agrobacterium tumefaciens mediated transformation.
Protoplast transformation:
Agrobacterium tumefaciens mediated transformation:
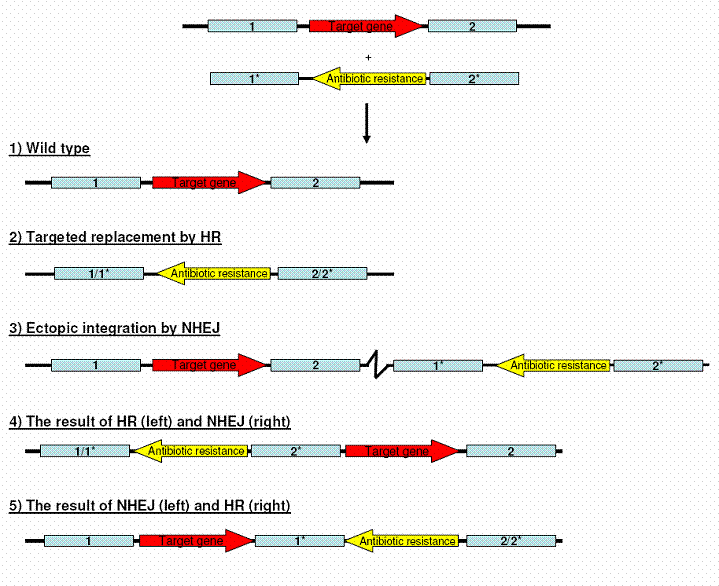
STEP 4: Homologous recombination between the introduced DNA and the genomic targetIn addition to the desired homologous recombination (HR) a competing mechanism exists, known as non-homologous end joining (NHEJ). This is also a DNA repair mechanism for dealing with double stranded DNA breaks; however it indiscriminately stitches free DNA ends together without ensuring that they fit together in an informative perspective. In plants and animals this repair mechanism is the most active one, possibly due to their very large genomes which makes it very unlikely to identify free DNA ends that fits together. In Fusarium graminearum approximately 60 % of the obtained antibiotic resistance transformants are the product of HR, while the remaining are a combination of NHEJ and NHEJ/HR. The figure below shows the five possible outcomes of a transformation. In situation 1 the vector DNA has not been taken up by the cell or has not been integrated into the genome. This is the dominating situation as transformation of 200.000 spores typically results in 100 to 200 antibiotic resistant transformants (0.005 – 0.01 % success rate). In situation 2 the target gene has been replaced by the antibiotic gene, which is the desired situation. In situation 3 the DNA has integrated randomly into the genome, leaving the target locus unchanged. This is the situation that is utilized for random mutagenesis and introduction of visual marker genes, such as green fluorescent protein (gfp). In situation 4 and 5 the DNA has been integrated into the target locus by HR in one end and NHEJ in the other end.
In addition to these situations comes different combinations due to the integration of multiple DNA copies.
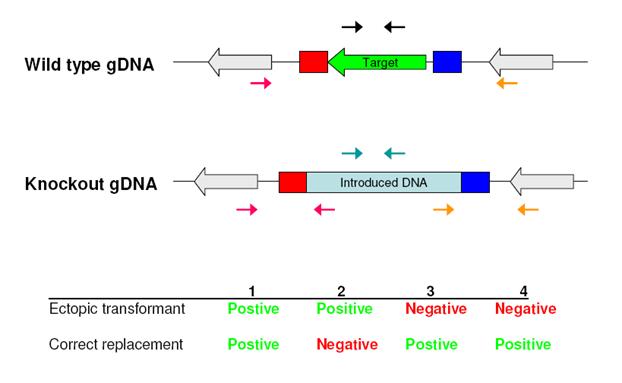
STEP 5: Identification and verification of replacement strainsIdentification and verification of replacement strains is a very important and often time consuming process. However, by combining microwave oven colony-PCR with a efficient PCR based screening strategy, correct transformants can be identified in a single workday. We use the following PCR primers.
This combination of primers allows us to distinguish between the five different transformant types, efficiently picking out correct transformants. However, the strategy does not sheath any light on the number of copies of the introduced resistance gene, which is relevant for transformants in which the target gene have been replaced and an additional vector DNA copy has been integrated ectopically in the genome. To reveal this rarely observed situation (for ATMT) a Southern blot is required.
Figure: Placement of the four primer pairs for verifying that homologous recombination has occurred correctly and the expected results.
|
|
Dette sted blev sidst opdateret 01. August 2010